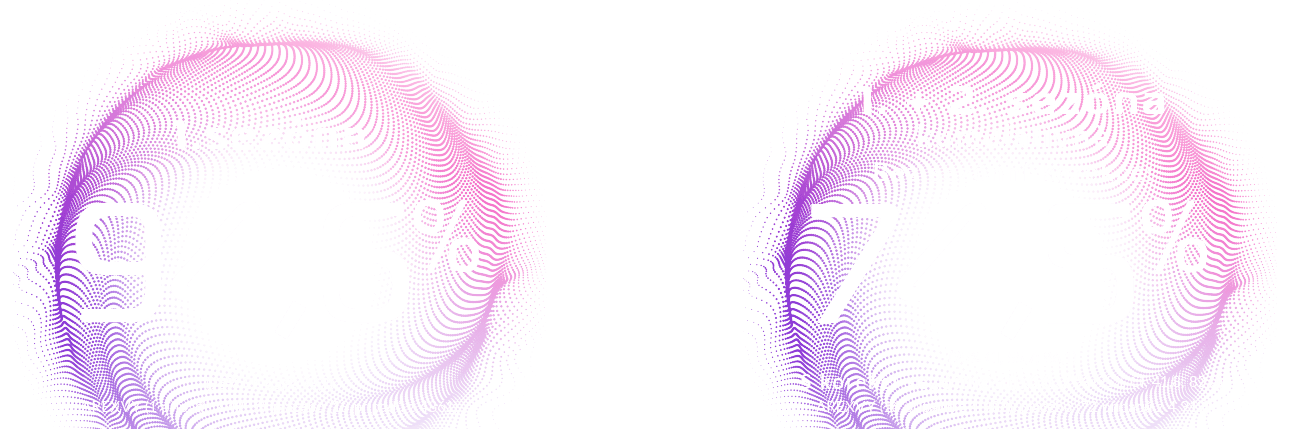DESIGN STUDIE1
Účinnost proti onemocněním dolních cest dýchacích (DCD) spojovaným s RSV u dospělých ve věku 60 let a starších byla hodnocena v rámci probíhající randomizované, placebem kontrolované, pro pozorovatele zaslepené klinické studie fáze III prováděné v 17 zemích severní a jižní polokoule. Účastníci budou sledováni po dobu až 36 měsíců.
Primární populace pro analýzu účinnosti (označovaná jako modifikovaný exponovaný soubor, zahrnující dospělé ve věku 60 let a starší, kteří obdrželi 1 dávku vakcíny Arexvy nebo placeba a u nichž do 15. dne po očkování nebylo hlášeno akutní respirační onemocnění (ARI) prokazatelně způsobené RSV) zahrnovala 24 960 účastníků rovnoměrně randomizovaných do dvou skupin, jimž byla podána 1 dávka vakcíny Arexvy (n = 12 466) nebo placeba (n = 12 494). V době primární analýzy účinnosti byli účastníci sledováni z hlediska rozvoje onemocnění DCD spojovaných s RSV po dobu 6,7 měsíce (medián).
Medián věku účastníků byl 69 let (rozmezí: 59 – 102 let), z čehož přibližně 74 % bylo starších 65 let, přibližně 44 % starších 70 let a přibližně 8 % starších 80 let. Přibližně 52 % byly ženy. Při vstupu do studie se u 39,3 % účastníků vyskytovala alespoň jedna komorbidita z oblasti zájmu studie; u 19,7 % účastníků bylo přítomno výchozí kardiorespirační onemocnění (CHOPN, astma, jakékoli chronické respirační/plicní onemocnění nebo chronické srdeční selhání) a u 25,8 % účastníků se vyskytovalo endokrinometabolické onemocnění (diabetes, pokročilé onemocnění jater nebo ledvin).
Primárním cílem bylo prokázat účinnost v prevenci první epizody onemocnění DCD prokazatelně spojeného s RSV-A a/nebo B v průběhu první sezóny po očkování. Potvrzené případy infekce RSV byly stanoveny pomocí kvantitativní polymerázové řetězové reakce s reverzní transkripcí (qRT-PCR) na výtěru z nosohltanu. Onemocnění DCD bylo definováno na základě následujících kritérií: účastník musel pociťovat alespoň 2 příznaky/známky postižení dolních cest dýchacích, přičemž alespoň 1 známka postižení dolních cest dýchacích musela být přítomna po dobu alespoň 24 hodin, nebo musel účastník pociťovat alespoň 3 příznaky postižení dolních cest dýchacích po dobu alespoň 24 hodin.
Příznaky postižení dolních cest dýchacích zahrnovaly: nový vznik nebo zvýšené množství sputa, nově se vyskytující nebo silnější kašel, nově se vyskytující nebo zvýšenou dyspnoe (dušnost). Známky postižení v dolních cestách dýchacích zahrnovaly: nově se vyskytující nebo zesílené pískoty, chrůpky/chrčení, dechovou frekvenci ≥ 20 dechů/min., nízkou nebo sníženou saturaci kyslíkem (saturace O2 < 95 % nebo ≤ 90 %, pokud je výchozí hodnota < 95 %) nebo potřebu podávání doplňkového kyslíku.
Imunogenita u dospělých ve věku 50 až 59 let se zvýšeným rizikem onemocnění RSV.
Non-inferiorita imunitní odpovědi na vakcínu Arexvy u dospělých ve věku 50 až 59 let ve srovnání s dospělými ve věku 60 let a staršími, u kterých byla účinnost vakcíny proti onemocnění DCD spojenému s RSV prokázána, byla hodnocena v pro pozorovatele zaslepené, randomizované, placebem kontrolované klinické studii fáze III.
Kohorta 1 zahrnovala účastníky ve věku 50 až 59 let rozdělené do 2 podskupin (dospělí se zvýšeným rizikem a dospělí bez zvýšeného rizika) na základě jejich anamnézy. Podskupina dospělých se zvýšeným rizikem zahrnovala účastníky s předem definovanými, stabilními, chronickými zdravotními stavy vedoucími ke zvýšenému riziku onemocnění RSV (Arexvy, n = 386; placebo, n = 191), jako je chronické plicní onemocnění, chronické kardiovaskulární onemocnění, cukrovka, chronické onemocnění ledvin nebo jater. Podskupina dospělých bez zvýšeného rizika sestávala z účastníků bez předem definovaných, stabilních, chronických zdravotních stavů (Arexvy, n = 383; placebo, n = 192).
Kohorta 2 (starší dospělí) zahrnovala účastníky ve věku 60 let a starší (Arexvy, n = 381).
Primárními cíli hodnocení imunogenity bylo prokázat 1 měsíc po vakcinaci non-inferioritu humorální imunitní odpovědi (z hlediska neutralizačních titrů RSV-A a RSV-B) po podání Arexvy u účastníků ve věku 50 až 59 let s předem definovanými, stabilními, chronickými zdravotními stavy vedoucími ke zvýšenému riziku onemocnění RSV a bez nich, ve srovnání s účastníky ve věku 60 let a staršími.


 Vaše registrace byla úspěšná!
Vaše registrace byla úspěšná! Váš účet byl aktivován – probíhá ověřování.
Váš účet byl aktivován – probíhá ověřování. Váš účet byl aktivován. Pokud by se nám v nejbližší době nepodařilo ověřit Vás jakožto odborníka ve zdravotnictví budeme Vás kontaktovat.
Váš účet byl aktivován. Pokud by se nám v nejbližší době nepodařilo ověřit Vás jakožto odborníka ve zdravotnictví budeme Vás kontaktovat. Nyní mate plný přístup ke všem stránkám a funkcím portálu GSK.
Nyní mate plný přístup ke všem stránkám a funkcím portálu GSK.